
Mount Sinai Medical Center
New York, New York
Chiari Malformations
What Are They?
Chiari malformations (CMs) are structural defects in the base of the skull and cerebellum. The cerebellum, and especially the cerebellar tonsils, are situated above the foramen magnum. With CMs, however, these parts of the brain extend into the upper spinal canal, often blocking the flow of cerebrospinal fluid (CSF).
There are several different causes of CMs. Often they are caused by structural defects in the brain and spinal cord that occur during fetal development, the result of genetic mutations, or a maternal diet lacking certain vitamins or nutrients (a primary or congenital CM). Also, CMs can be caused—although much less commonly—later in life if the CSF is drained excessively from the lumbar or thoracic areas of the spine, either due to traumatic injury, disease, or infection (an acquired or a secondary CM). Obstruction of the flow of CSF may cause a syrinx to form, eventually leading to syringomyelia.
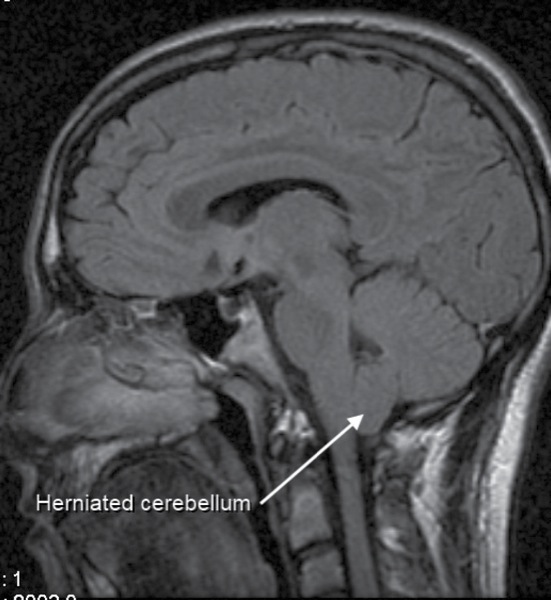
Chiari malformations are further classified by the severity of the disorder and the parts of the brain that protrude into the spinal canal.1
In Type I, the cerebellar tonsils extend into the foramen magnum. This condition may be asymptomatic and is the most common form of CMs, usually first noticed in adolescence or adulthood, and often is found coincidentally during an examination for another condition. Patients with asymptomatic CMs may develop signs of the disorder later in life.
Individuals with Type II or classic CMs have symptoms that are generally more severe than those seen in Type I. Life-threatening complications requiring urgent surgical correction may appear during childhood. Both the cerebellum and brain stem protrude into the foramen magnum. The midline vermis that connects the two halves of the cerebellum may be missing or only partially formed. Type II is usually accompanied by a myelomeningocele or spina bifida. The term Arnold-Chiari malformation—named after Austrian pathologist Hans Chiari and German pathologist Julius Arnold—is specific to CM Type II.
Type III is very rare and the most serious malformation. Parts of the cerebellum and the brain stem herniate through an abnormal opening in the back of the skull. Symptoms of Type III appear in infancy and cause debilitating and life-threatening complications. Mental and physical delays as well as seizures are common.
Type IV also is rare, and although the cerebellum is located in its normal position, parts are missing or incomplete (cerebellar hypoplasia). Portions of the skull and spinal cord may be visible.
Incidence
The prevalence of tonsillar herniations of 3 to 5 mm or greater (CM Type I) was previously believed to be about 1 per 1,000 births, but is likely much higher as the syndrome often goes undetected.2 The male:female incidence is 1:3. Type II malformations are more common in people of Celtic descent, and mortality without surgery is high. Cerebellar tonsillar ectopia has been diagnosed during upright MRI in 23% of adults with headache following motor vehicle head trauma.3 Evolutionary and genetic factors are also involved. Normally the posterior fossa makes up 27% of the total intracranial space, whereas in adults with CM Type I, it accounts for only 21%.4
Diagnosis
Diagnosis requires patient history, neurologic examination, and medical imaging. MRI is the preferred imaging modality, as it visualizes the cerebellar tonsils and spinal cord as well as bone and other soft tissues. CT and CT myelography also are options. Diagnosis of a CM Type II can be made prenatally through ultrasound.
In older patients, common symptoms include headaches, difficulty swallowing, choking and vomiting, dizziness, poor gag reflex, nausea, neck pain, unsteady gait, poor hand coordination, nystagmus, papilledema, papillary dilation, facial pain, impaired fine motor skills, numbness and tingling of the hands and feet, tinnitus, cardiac arrhythmias (tachycardia or syncope), L’Hermitte’s sign (tingling that runs down the back and into the limbs), central sleep apnea and other abnormal breathing patterns, speech difficulties, and even paralysis. Other conditions occasionally associated with CM include hydrocephalus, syringomyelia, spinal curvature, tethered spinal cord syndrome, and connective tissue disorders (Ehlers-Danlos and Marfan syndromes). Headaches can be aggravated by Valsalva maneuvers, such as yawning, laughing, crying, coughing, sneezing or straining, bending over, or getting up suddenly.4
Treatment
There is no known cure. Treatments include surgery (decompressive craniectomy) and management of symptoms, based on the occurrence of clinical symptoms rather than radiological findings. Decompressive surgery requires removal of the lamina of the first and often the second or third cervical vertebrae and part of the occipital bone of the skull to relieve pressure. A shunt may be required to increase CSF flow. A dural graft is applied to cover the expanded posterior fossa. In some cases, detethering the spinal cord may be used as an alternate approach to relieve compression of the brain against the foramen magnum.
Complications from decompression surgery can arise. Surgery is usually done in the prone position. Rarely, compression of the brain stem occurs anteriorly. In these cases, an anterior decompression is required. The approach is done transorally—tracheostomy is performed after induction and endotracheal intubation—to remove the bone compressing the brain stem, typically the odontoid.
Anesthetic Precautions
Documentation of the physical condition is essential, including airway status, cardiac assessment, and neurologic status. If the patient suffers from gastrointestinal problems, such as vomiting, prophylaxis is recommended. The presence of sleep apnea should be appreciated and appropriate measures enacted. Care of the newborn requires careful attention to temperature and infection control. Precautions for the prone position include attention to appropriate padding, attention to eye protection, careful fluid management to decrease airway edema, and the risk for postoperative visual loss and nerve damage.
The accessory nerve (CX1) is especially sensitive to damage during surgery. Electromyographic recording is usually maintained intraoperatively. Muscle relaxants should be used sparingly or not at all during the period of dissection. As soon as possible in recovery, the ability to shrug the shoulders should be documented, as CX1 supplies the sternocleidomastoid and trapezius muscles. If other neuromonitoring (somatosensory) is used, inhalation agents should be used in conjunction with IV agents. Multimodality intraoperative neurophysiologic monitoring has been found to be valuable in pediatric populations, although less so in adults.5,6 Accurate intraoperative documentation is important. Normotension is better achieved with vasopressors than fluid boluses.
The aim for laboring patients is to reduce straining and pain during contractions that might increase intracranial pressure, headache, and herniation. A literature review found that general anesthesia and neuraxial blocks can be used safely without fetal damage or worsening of maternal symptoms postpartum.7-9
Moyamoya Disease

What Is It?
Moyamoya disease is a progressive, occlusive disease of the cerebral vasculature with particular involvement of the circle of Willis, the basal ganglia, and the arteries that feed these areas.10 To compensate for the obstructed vessels, tangles of smaller vessels develop to maintain blood flow. Although the cause is unknown, the disease is believed to be genetic. The first gene identified was RNF213. Susceptible loci have been found on 3p, 6p, 17q, and band 8q23. Other loci are q25.3 on chromosome 17 with a strong association between p.R4859K and p.R4810K and polymorphisms of the RNF213 gene.11-13 Immune abnormalities also may occur, as there is a high incidence of elevated thyroid antibodies. Distinct genetic backgrounds exist in addition to environmental factors. Ethnicity-specific factors indicate complex determiners based on country of origin.
Incidence
In one study, the prevalence of Moyamoya disease in California and Washington was 0.086 cases per 100,000 people.14 The incidence was higher in whites than Hispanics. However, in Japan the incidence is reported to be 0.35 cases per 100,000, with a prevalence of 3.16. The ratio of women to men affected is estimated at 1.8:1. Age at first diagnosis ranges from 6 months to the seventh decade, with the highest peak in the first decade.15
Diagnosis
Diagnosis is made by CT, MRI, or magnetic resonance angiography. Single-photon emission CT demonstrates decreased blood and oxygen supply to involved cerebral areas. Angiography provides the conclusive diagnosis.
Clinical presentations differ widely, from no symptoms to cerebral hemorrhage and multiple strokes. Hemorrhage, often of sudden onset, is more common in adults, and cerebral ischemic events affect children more frequently, manifesting as hemiparesis, sensory impairments, involuntary movements, seizures, and headaches. Mental retardation may be progressive.
Although Moyamoya disease may occur as a single disorder, many other disease states have been associated with it: skin abnormalities; dysmorphism; thyroid malfunction; leptospirosis; tuberculosis; aplastic and sickle cell anemia; neurofibromatosis; multiple vascular disorders; and Down, Apert, and Marfan syndromes; as well as other disorders. When another disorder has been identified in conjunction with Moyamoya disease, the condition is known as Moyamoya syndrome.
Treatment
No cure is known. Antiplatelet agents, including aspirin, may be prescribed to prevent clots. Surgery is usually recommended to direct extracranial flow intracranially (eg, superficial temporal artery to the middle or anterior cerebral artery [STA-MCA] or external carotid to internal carotid bypass). Many operations have been developed, including EDAS (encephaloduroarteriosynangiosis), a synangiosis that is essentially the same as STA-MCA; and EMS (encephalomyosynangiosis), where the temporalis muscle is passed through the skull and placed on the surface of the brain, and multiple small burr holes are made to allow growth from new vessels into the brain from the scalp.16
Anesthetic Precautions
Review and documentation are essential due to the many associated conditions. Airway and neurologic status are of particular importance. Several reviews have examined the effects of different anesthetic techniques.17-20 In a pediatric study of 22 children, 1 death and 4 new postoperative neurologic complications were associated with female gender and high opioid dose.17 Another study examined the perioperative course of 15 patients undergoing revascularization procedures. The type of anesthesia used for induction and maintenance did not appear to affect outcome; however, either low end-tidal carbon dioxide (<31 mm Hg) or high levels (>35 mm Hg) significantly prolonged hospital stay.18
Because the intracranial pressure during labor is sensitive among Moyamoya parturients, adequate anesthesia and maintenance of hemodynamic status are essential. Literature review indicates that cesarean delivery and uncomplicated vaginal delivery under epidural anesthesia have been used successfully.19 Avoidance of hypotension postoperatively due to autonomic dysfunction also is important, as an awake patient may suddenly become comatose due to cerebral ischemia.20
References
- Hydrocephalus. https://emedicine.medscape.com/?article/?1135286-overview. Accessed December 29, 2017.
- Milhorat TH, Bolognese PA, Nishikawa M, et al. Syndrome of occipitoatlantoaxial hypermobility, cranial settling, and Chiari malformation type I in patients with hereditary disorders of connective tissue. J Neurosurg Spine. 2007;7(6):601-609.
- Freeman MD, Rosa S, Harshfield D, et al. A case-control study of cerebellar tonsillar ectopia (Chiari) and head/neck trauma (whiplash). Brain Inj. 2010;24(7-8):988-994.
- Furtado SV, Reddy K, Hegde AS. Posterior fossa morphometry in symptomatic pediatric and adult Chiari I malformation. J Clin Neurosci. 2009;16(11):1449-1454.
- Barzilai O, Roth J, Korn A, et al. The value of multimodality intraoperative neurophysiological monitoring in treating pediatric Chiari malformation type I. Acta Neurochir (Wien). 2016;158(2):335-340.
- Roser F, Ebner FH, Liebsch M, et al. The role of intraoperative neuromonitoring in adults with Chiari I malformation. Clin Neurol Neurosurg. 2016;150:27-32.
- Garvey GP, Wasade VS, Murphy KE, et al. Anesthetic and obstetric management of syringomyelia during labor and delivery: a case series and systematic review. Anesth Analg. 2017;125(3):913-924.
- Choi CK, Tyagaraj K. Combined spinal-epidural analgesia for laboring parturient with Arnold-Chiari type I malformation: a case report and a review of the literature. Case Rep Anesthesiol. 2013;2013:512915.
- Ghaly RF, Tverdohleb T, Candido KD, et al. Management of parturients in active labor with Arnold Chiari malformation, tonsillar herniation, and syringomyelia. Surg Neurol Int. 2017;8:10.
- Scott RM, Smith ER. Moyamoya disease and Moyamoya syndrome. N Engl J Med. 2009;360(12):1226-1237.
- Liao X, Deng J, Dai W, et al. Rare variants of RNF213 and Moyamoya/non-Moyamoya intracranial artery stenosis/occlusion disease risk: a meta-analysis and systematic review. Environ Health Prev Med. 2017;22(1):75.
- Mineharu Y, Liu W, Inoue K, et al. Autosomal dominant moyamoya disease maps to chromosome 17q25.3. Neurology. 2008;70(24 pt 2):2357-2363.
- Kamada F, Aoki Y, Narisawa A, et al. A genome-wide association study identifies RNF213 as the first Moyamoya disease gene. J Hum Genet. 2011;56(1):34-40.
- Uchino K, Johnston SC, Becker KJ, et al. Moyamoya disease in Washington state and California. Neurology. 2005;65(6):956-958.
- Kim JS. Moyamoya disease: epidemiology, clinical features, and diagnosis. J Stroke. 2016;18(1):2-11.
- Gonzalez NR, Dusick JR, Connolly M, et al. Encephaloduroarteriosynangiosis for adult intracranial arterial steno-occlusive disease: long-term single-center experience with 107 operations. J Neurosurg. 2015;123(3):654-661.
- Jagdevan S, Sriganesh K, Pandey P, et al. Anesthetic factors and outcome in children undergoing indirect revascularization procedure for Moyamoya disease: an Indian perspective. Neurol India. 2015;63(5):702-706.
- Samagh N, Bhagat H, Grover VK, et al. Retrospective analysis of perioperative factors on outcome of patients undergoing surgery for Moyamoya disease. J Neurosci Rural Pract. 2015;6(2):262-265.
- Weston E, Mehta N. Moyamoya disease in pregnancy and delivery planning: a case series and literature review. Obstet Med. 2016;9(4):177-180.
- Machida T, Higuchi Y, Nakano S, et al. Postoperative cerebral ischemia due to hypotension in Moyamoya patient with autonomic dysfunction. World Neurosurg. 2018;109:204-208.