
Department of Anesthesiology and Perioperative Medicine
Penn State College of Medicine
Hershey, Pennsylvania
Partner, Riverside Anesthesia Associates
Staff Anesthesiologist
UPMC Pinnacle Hospital System
Harrisburg, Pennsylvania
Pain is not only difficult to treat but difficult to understand because it involves many complex processes that lead first to its recognition and then to an emotional response and reaction. As anesthesia providers, our interest in pain continues to grow because most of us will, at some point, be challenged by significant perioperative pain, and 100% of these patients will require treatment. Furthermore, concerns about the overuse of opioids for acute and chronic pain militate for a reduction in their use, and so anesthesiologists strive to find effective alternatives.
First, we need to understand that pain and nociception are two different things. Pain is the unpleasant experience caused by potential or actual tissue damage, and nociception is the transmission of a noxious stimulus to the brain and all the processes in between.
The goal of this article is to methodically but simply walk through the pain pathway as it is currently understood, and show where therapies, both pharmacologic and physical, can intervene.
The 4 Steps of the Pain Pathway: Transduction, Transmission, Modulation, and Perception
Transduction: How a Mechanical Stimulus in Tissues Becomes an Electrical Signal in Nerves
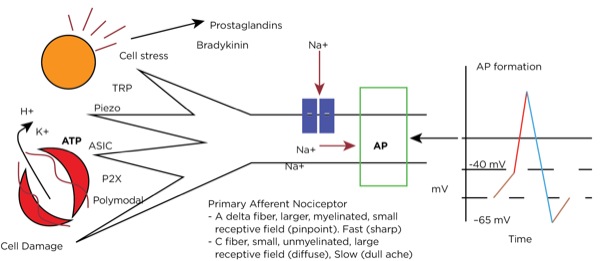
Transduction occurs when an insult to our tissues, such as a nail piercing the foot, a cut from a surgeon’s scalpel, or an infectious process, is converted to an action potential in a primary afferent neuron. When there is potential or actual tissue damage, substances such as prostaglandins are produced. These inflammatory mediators either directly stimulate the nociceptors (pain receptors in the tissues) or sensitize them to more readily accept the noxious stimulus. Furthermore, when tissue damage occurs, potassium (K+), adenosine triphosphate (ATP), and hydrogen ions from the cells directly stimulate the nociceptors.
Nociceptors can be external, and so are found in the skin at varying densities based on location (higher concentrations in the fingertips, hands, and face, and lower concentrations over the torso). They can also be internal, and so are within muscles, joints, bones, and internal organs. There are specific receptors for different stimuli—such as Piezo receptors for mechanical stimuli, transient receptor potential (TRP) receptors for extremes in temperature, P2X purinergic receptors for ATP, and acid-sensing ion channels (ASICs) for hydrogen ions—or polymodal nociceptors that can be activated by multiple types of stimuli.
The primary afferent nerves that contain these nociceptors are either A delta fibers, which are large and myelinated and responsible for acute sharp pain, or C fibers, which are small and unmyelinated and responsible for slow-onset, dull, lingering, or achy pain. Stimulation of the nociceptors’ open voltage-gated ion channels allow calcium and sodium ions to pass into the cytoplasm, raising the resting membrane potential (around –65 mV) downstream until the threshold potential (around –40 mV) is achieved, leading to action potential formation. (Figure 1.)
Transmission: Propagation of the Electrical Signal From Nerves to the Brain
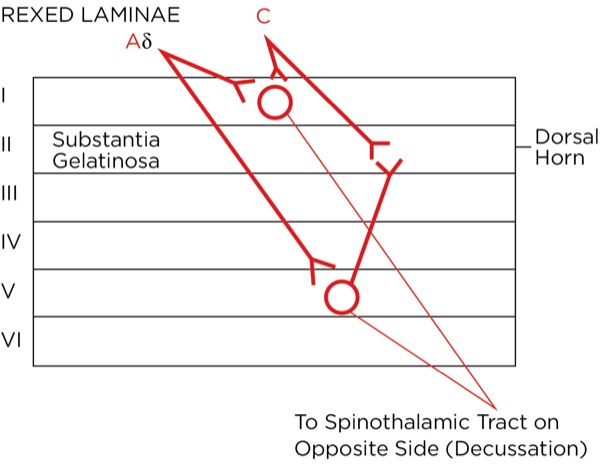
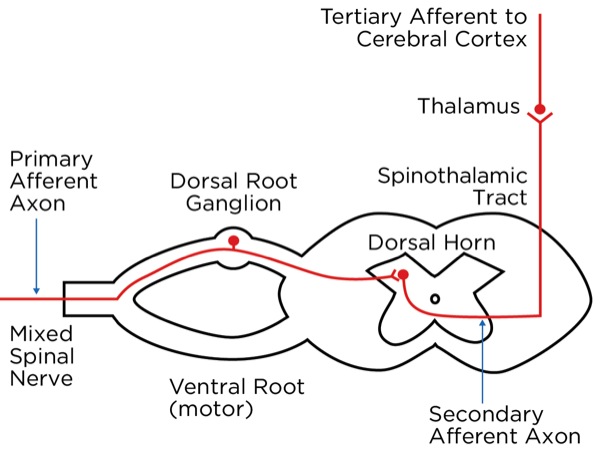
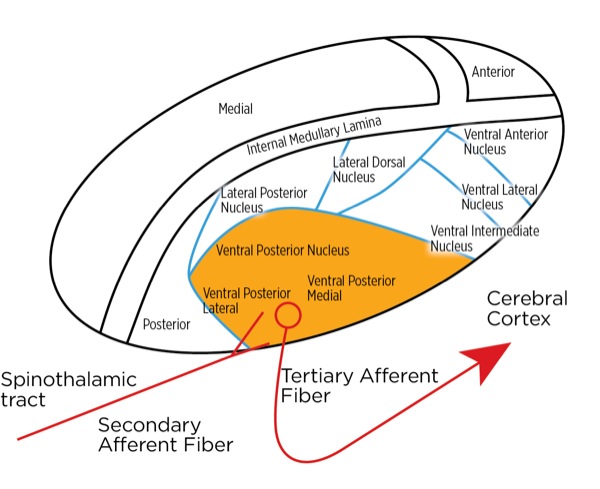
The signal as an action potential travels up the primary afferent axon as it is propagated by the continued reaching of threshold potential due to the opening of voltage-gated Na+ channels upstream (termed saltatory conduction). The primary afferent neurons have their cell bodies in the dorsal root ganglion. The primary afferent neurons synapse with secondary efferent neurons in the dorsal horn of the spinal cord (Rexed laminae I, II, or V) (Figure 2). The action potential generated in the secondary afferent neuron then crosses over to the other side of the spinal cord (decussation) within a few levels of the stimulus and ascends primarily within the spinothalamic tract (Figure 3). The secondary afferent neurons synapse with tertiary afferent neurons in the thalamus (which acts as the relay station between the brain and the rest of the nervous system; Figure 4), and the action potential generated in the tertiary afferent neuron then travels primarily to the somatosensory cortex (Figure 5).
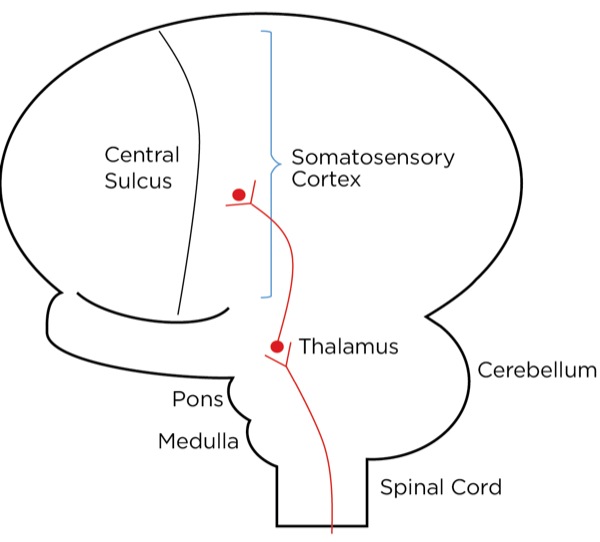
Other cortical areas receiving input include the anterior cingulate cortex, the insular cortex, the ventrolateral orbital cortex, and the motor cortex. Together, they localize the pain and orchestrate an emotional, autonomic, and motor response. The neurotransmitters commonly involved with the ascending pathway are glutamate (primary) and substance P (secondary) within the spinal cord; others, including gamma-aminobutyric acid (GABA), norepinephrine, serotonin, dopamine, and endogenous opioids, are involved within the cortexes.
Within the dorsal horn, primary afferent neurons from the viscera (heart, etc) synapse with the secondary afferent neurons as well. Since the visceral primary afferents are usually “silent,” their action potentials are frequently interpreted by the cortexes as signals coming from other “commonly” active primary afferent neurons within the same part of the body. This leads to “referred pain.”
Modulation: Altering or Blocking the Pain Signal as It Travels Through Spinal Cord, Medulla, Pons, and Midbrain to the Cerebral Cortex
Modulation is the way the brain (descending pathway) alters the intensity of the signal traveling up the ascending pathway depending on the circumstance surrounding the initiation of the nociceptive signal.
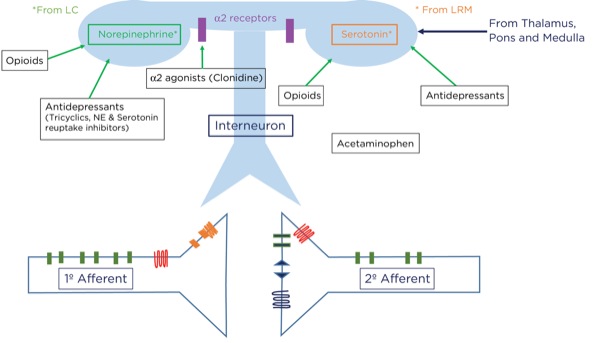
For example, if you sprain your ankle while running from a lion, it would be best to be able to ignore the pain and keep running. How does this occur? Signals originating in the cerebral cortex as well as the spinothalamic tract pass through the periaqueductal gray matter in the midbrain. A signal as an action potential is generated there which then travels to the pons (locus coeruleus) and medulla (locus raphe magnus) (Figure 6). From there, another action potential is generated that travels down the spinal cord through the dorsolateral tract, terminating upon an interneuron near the synapse between the primary and secondary afferent neurons.

At that level, inhibitory signals are sent by the interneuron, which alters or inhibits (modulates) the pain signals traveling to the brain by decreasing the release of glutamate and substance P from the presynaptic terminals of the primary afferent axons and reducing the postsynaptic excitatory signals originating in the postsynaptic terminal of the secondary afferent axons. Many neurotransmitters are involved here as well, including norepinephrine, serotonin, and primarily endogenous opioids (enkephalins).
This is part of the gate theory, proposed by Melzack and Wall in 1965.1 This theory helps to explain why transcutaneous electrical nerve stimulation (TENS) units work, or rubbing a painful area soothes the pain. By providing other nonpainful signals via non-nociceptive afferent neurons (the much larger, myelinated A beta fibers), the site of the synapse between the primary and secondary afferent neurons becomes busy with incoming signals which creates a sort of traffic jam, slowing or blocking the action potentials triggered by the activation of the nociceptors at the site of injury (Figure 7).
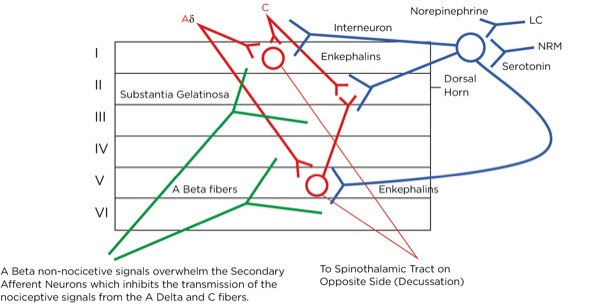
Perception: How the Brain Interprets the Signal and Produces “Pain”
Perception occurs when the nociceptive signal is received by the involved cortexes within the brain. The individual becomes aware of the insult, and an emotional and motor response is initiated. It has reached consciousness and now moves from nociception to pain.
Inflammation and Inflammatory Pain
When tissues are damaged, inflammatory mediators are released, resulting in arteriole dilation that then causes the area to become red and hot (rubor and calor). Furthermore, the endothelium of capillaries and venioles contracts, opening spaces for fluid and cells to escape into the inflamed area, which causes swelling (tumor). As noted below, these same mediators cause pain (dolor). The nociceptors can eventually become sensitized to the signals they receive, causing allodynia (pain from nonpainful stimuli) and hyperalgesia (exaggerated pain from a painful stimulus).
Arachidonic acid is freed from intracellular phospholipids when tissues are damaged or there is a threat of tissue damage. Other substances are then formed from arachidonic acid, most notably prostaglandins, which are produced from its breakdown by cyclooxygenase-2 (COX-2), which is produced in high concentrations during inflammation. The prostaglandins are important to perpetuate the inflammatory process (PGI2 or prostacyclin), and trigger and sensitize nociceptors (prostaglandin E2 [PGE2]).
How do the inflammatory mediators sensitize the nociceptors? By PGE2 binding to specific areas on the nociceptors on A delta and C fibers (creating cyclic AMP from ATP), and by altering voltage-gated sodium channels, the depolarization threshold is lowered, meaning minimal nonpainful stimuli cause a nociceptive action potential to be produced (allodynia) and painful stimuli to trigger more action potentials (hyperalgesia). Furthermore, other receptors, such as transient receptor potential vanilloid 1 (TRPV1), which is sensitive to high temperatures, will be altered to fire at lower temperature levels, thus creating temperature-dependent allodynia. The C fiber nociceptors can be altered to the point that the threshold potential is at resting potential, so action potentials are continuously produced, creating ongoing unrelenting pain during inflammation.
How is bradykinin formed during inflammation? Hageman factor (factor XII), high-molecular-weight (HMW) kininogen, and prekallikrein are produced in the liver and released into the bloodstream. When they pass into the inflammatory exudate, factor XII is converted to activated factor XII (factor XII A) when it interacts with substances such as collagen. Factor XII A converts prekallikrein to kallikrein. Kallikrein in turn converts HMW kininogen to bradykinin. Bradykinin, like PGE2, is important in the inflammatory process because it too directly stimulates and sensitizes peripheral nociceptors. Furthermore, as with PGE2, it sensitizes the TRPV1 receptor, creating thermal allodynia and hyperalgesia.
Analgesics
NSAIDs
NSAIDs, such as aspirin and ibuprofen, block the synthesis of all the prostaglandins including PGI2 (prostacyclin), thereby slowing or stopping the inflammatory process and reducing pain by blocking the formation of PGE2.2 They do this by blocking cyclooxygenase-1 (COX-1) and COX 2. COX-1 is found in low concentrations at steady state throughout the body, and COX-2 is the inducible form, which is dramatically increased during inflammatory states.
Glucocorticoids
The glucocorticoids, such as hydrocortisone and dexamethasone, reduce inflammatory pain by blocking the intracellular conversion of phospholipids to arachidonic acid, thereby not only inhibiting the production of prostaglandins but also leukotrienes (their production is not blocked by NSAIDs). Furthermore, the glucocorticoids, which are corticosteroids, work within the nucleus of the cell to decrease the transcription of proinflammatory genes and increase the transcription of anti-inflammatory genes.3 Glucocorticoids have also been shown to reduce spontaneous action potential firing in injured nerves, both centrally and peripherally (Figure 8).
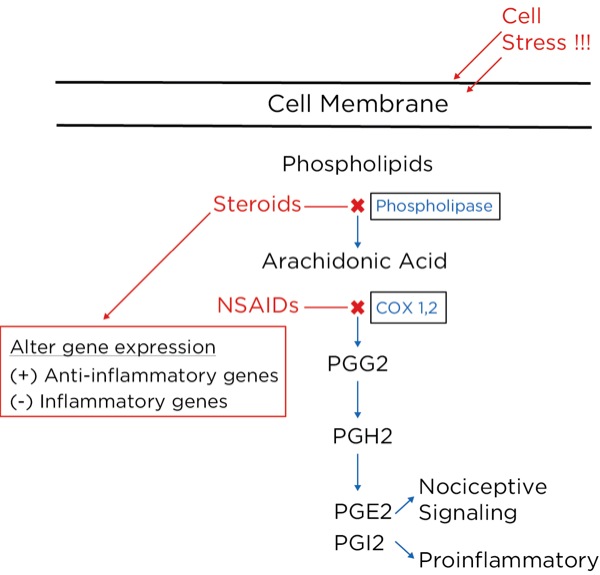
Acetaminophen
Acetaminophen has been grouped with the NSAIDs because it is a COX inhibitor; however, it appears to be effective only when levels of inflammatory mediators are very low, making it ineffective when there is a significant amount of inflammation. Acetaminophen is, however, an analgesic. It has been suggested that acetaminophen works by blocking COX-3, which is a splice variant of COX-1, found within the central nervous system, and possibly lowering concentrations of nitric oxide (NO); that mechanism, however, is not completely understood.5 There is evidence that it activates serotonergic pathways, thereby potentiating pain modulation.
Opioids
Opioids bind to and stimulate opioid receptors (mu, kappa, and delta) found throughout the brain, spinal cord, peripheral nervous system, and GI tract. To understand how opioids such as morphine work, we need to have a basic understanding of what opioids do when they bind to an opioid receptor.
The opioid receptors are a type of G protein–coupled receptor (GPCR). GPCRs are found throughout the central and peripheral nervous system and are involved in many aspects of nervous system function, including sympathetic and parasympathetic activity and pain modulation. The GPCR passes through the cell membrane 7 times, creating 3 extracellular and 3 intracellular loops. There is an extracellular N-terminus and intracellular C-terminus. The extracellular loops and N-terminus create the pocket that binds the ligand, such as an opioid. The receptor is bound intracellularly to a heterotrimeric (3 subunits) G protein consisting of alpha, beta, and gamma subunits, bound together while in the inactive state. While inactive, the subunit has guanosine diphosphate (GDP) bound to it. With the ligand binding to the receptor, a conformational change occurs within the alpha subunit, leading to the release of the GDP and the binding of guanosine triphosphate (GTP). This then leads to a separation of the alpha subunit from the beta and gamma subunits, which stay bound together. Separately, the 2 subunits (alpha and beta-gamma) are active and promote intracellular signaling (Figure 9).
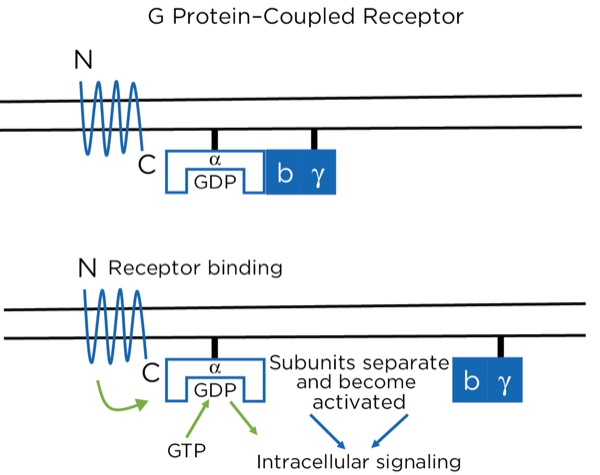
With the opioids, the intracellular signaling involves potentiating or reducing the production of cyclic adenosine monophosphate (cAMP). At certain sites, it signals the closing of voltage-gated Ca+ channels and the opening of K+ channels, leading to intracellular hyperpolarization of the neuron. At the synapse between the primary and secondary afferent neurons, for example, this leads to a reduction in the release of neurotransmitters presynaptically and a reduction in action potential production postsynaptically.
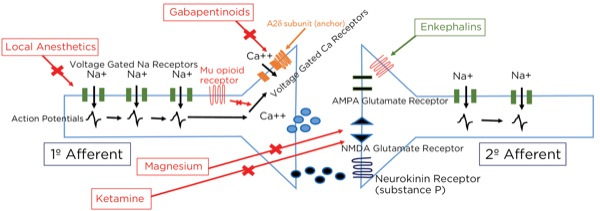
Where are these opioid receptors? As it pertains to pain modulation, as stated previously, some signals from the sensory cortex or from the afferent fibers that travel through the spinothalamic tract terminate in the periaqueductal gray of the midbrain. Endogenous opioids (enkephalins) are released that stimulate the neurons with fibers traveling to the locus coeruleus within the pons and the locus raphe magnus in the medulla. Enkephalins are released there as well, which stimulate the neurons with axons that eventually terminate upon the interneurons within the dorsal horn in the Rexed laminae II (substantia gelatinosa). The fibers from the locus coeruleus release norepinephrine and those from the locus raphe magnus release serotonin.
The interneurons send fibers that terminate at the synapse between the primary and secondary afferent fibers. When stimulated, the interneurons also release enkephalins, which decrease the release of glutamate and substance P presynaptically and decrease the excitatory current postsynaptically, thereby lessening or inhibiting the pain signal traveling to the brain.
Because exogenous opioids, such as morphine and fentanyl, perform the same action as the enkephalins at the receptor level, they can have an effect anywhere that enkephalins work—especially the midbrain, pons, medulla, and spinal cord.
Local Anesthetics
Action potentials are propagated by the opening of sodium (Na+) channels within the axons reaching threshold potentials downstream by saltatory conduction. Na+ channels exist in 3 phases: closed, open, and inactive. The inactive state is when the segment of axon is still depolarized but sodium cannot pass through, and its structure is distinctly different from the closed state.
Local anesthetics can only block the channels when they are in the open or inactive states, and they block the receptors from within the cytoplasm, meaning the drug must pass into the cell to be effective. Only the un-ionized form can pass through the cell membrane; therefore, as a weak base there is less available drug within the cytoplasm in an acidic environment. This is why local anesthetics are less effective in inflamed or infected tissues.
Another interesting phenomenon when using local anesthetics, based on their mechanism of action, is that they are much more effective within axons with more frequent action potentials, such as after a painful stimulus (or the chaotic signals of an arrhythmia). This is because the slower arrival of signals gives time for the local anesthetic molecule to be removed and the channel to revert to the closed state, ready for the next action potential. With large doses of local anesthetics, even slow-arriving signals will be blocked, leading to cardiac and CNS toxicity. Also, local anesthetics are more effective on smaller and unmyelinated neurons such as A delta and C fibers (compared with A beta fibers), which are most likely to be passing nociceptive signals.
Magnesium
As noted previously, glutamate is the primary neurotransmitter at the synapse between the primary and secondary afferent axons in the spinal cord dorsal horn. The glutamate can bind to 2 different receptors at the postsynaptic endplate: AMPA type and NMDA type. Even though glutamate binds to it, the NMDA-type receptor will not usually allow passage through the channel because it is blocked by magnesium. Magnesium is held there by the electrical gradient between the extracellular space and the intracellular space, with the intracellular space being more negative (–65mV). With painful stimuli, there is an increase in glutamate release and AMPA-type receptor binding due to more frequent action potentials, which are produced by more frequent triggering of the peripheral nociceptors. The electrical gradient across the cell membrane is reduced and the magnesium pops off, allowing for the opening of the NMDA-type receptors and the winding up of the nociceptive signals passing through the secondary afferent axons. By giving magnesium, more molecules are available to keep the NMDA-type receptors blocked during increased activity.
Magnesium is also a direct muscle relaxant and reducer of muscle pain since it is a promotor of a reduction in cytoplasmic calcium levels by stimulation of calcium reuptake into the sarcoplasmic reticulum, and it competes with calcium for plasma protein-binding sites.
Alpha-2 Agonists
The primary mechanism of action for the analgesic effects of the alpha-2 agonists is thought to involve modulation at the dorsal horn by binding to the postsynaptic alpha receptors of the interneurons. As noted previously, stimulation of these interneurons promotes the release of endogenous opioids.
Furthermore, analgesia may be produced by reducing peripheral sympathetic tone and reducing the responsiveness, or perception, within the brain by binding to presynaptic alpha-2 receptors, thereby reducing the release of catecholamines.
Ketamine
Ketamine likely exerts its analgesic effect primarily through the blockade of NMDA receptors throughout the central nervous system, thereby decreasing brain excitation and nociceptive signal sensitization, such as reducing the windup phenomenon by blocking post-synaptic NMDA-type glutamate receptors within the dorsal horn. Secondarily, ketamine decreases reuptake of serotonin and norepinephrine, thereby increasing neurotransmitter availability within the descending modulation pathway, and has opioid agonist activity. Its mood stabilization effect may evoke altered pain perception as well.
Antidepressants
The antidepressants are a diverse group of molecular structures and include those that are commonly used for pain management: the tricyclics (eg, amitriptyline, nortriptyline, doxepin) and serotonin and norepinephrine reuptake inhibitors (SNRIs, eg duloxetine). Selective serotonin reuptake inhibitors (SSRIs; eg, fluoxetine, sertraline) have been used for pain management but appear to be less effective than the other classes. All antidepressants have the ultimate effect of increasing excitatory neurotransmitters, such as norepinephrine and serotonin, in the synaptic cleft. Tricyclic antidepressants and SNRIs achieve this effect by blocking the reuptake of the neurotransmitters at the presynaptic terminals. Increasing the availability of norepinephrine and serotonin within the dorsal horn, thereby inhibiting pain transmission, is thought to be the primary modality for pain suppression. Other mechanisms, such as a direct effect on mu opioid receptors and improving pain perception by mood stabilization, have been postulated.5
Gabapentinoids
Although the gabapentinoids (gabapentin and pregabalin [Lyrica, Pfizer]) have a similar structure to GABA, they are not GABA agonists and do not increase GABA levels.6 The gabapentinoidstarget the alpha 2 delta-1 (A2delta-1) subunits of the voltage-gated calcium channels (VGCCs), predominantly within the dorsal horn of the spinal cord. When stimulated, the VGCCs allow calcium influx into the presynaptic terminals, leading to the mobilization and release of neurotransmitters. The A2delta-1 subunits are important for the stability of the VGCCs at the cell surface, and their uncoupling leads to the internalization and degradation of the channels, with a decrease, therefore, in the intracellular movement of calcium and release of the neurotransmitter glutamate.
The VGCCs are upregulated within damaged neurons as an adaptive response leading to an exaggerated influx of calcium and neurotransmitter release, with the arrival of an action potential, leading to neuropathic pain, demonstrating the obvious benefit of the administration of gabapentinoids for treating neuropathy. However, their effectiveness for treating acute pain is less obvious. This class of drugs is likely not capable of being the sole modality for treating acute surgical pain, and the effect is known to be present but limited, which is consistent with the above-described mechanism of action and known upregulation with nerve damage, but not acute tissue injury. Other mechanisms involving NMDA receptor blockade, inhibition of Na+ currents, and increased serotonin and norepinephrine activity leading to stimulation of the descending pain modulation pathway have been described (Figures 10 and 11).
Capsaicin
Capsaicin is a vanilloid and binds to the TRPV1 receptor of the primary afferent nociceptor.7 This is the receptor that responds to painful high temperatures. The binding of the receptor initially causes a depolarization and thus nociceptor activation, leading to intense, burning pain. After the initial depolarization, the TRPV1 receptor is deactivated, there is a continued hyperpolarization at the site, and then a temporary dying-back of the nerve endings, allowing the area to become refractory to all nociceptive stimuli and leading to a prolonged analgesic effect.
Nonpharmacologic Modalities
Placebo Effect: The Power of Higher Brain Centers For Pain Suppression
As an introduction to nonpharmacologic treatment, an understanding of the effectiveness of placebo is warranted. Over the years, it has been difficult for a new analgesic with a known acceptable mechanism of action to gain FDA approval within the United States due to an inability to demonstrate superiority over placebo. Studies have concluded that it is not necessarily due to the ineffectiveness of the drug but the power of the placebo effect. Placebo may be effective greater than 40% of the time, and it may be responsible for over 50% of the effect of a known pain reliever.8 Cortical sites as well as the periaqueductal gray and the descending pain modulation pathway are activated with the expectation of pain relief, a process that can be at least partially reversed by naloxone (opioid antagonist), implying that endogenous opioids are involved. A physiological as well as a psychological etiology is, therefore, implicated.
Other Modalities
The use of nonpharmacologic approaches to treat pain has been employed in acute and chronic pain settings.
Patient education has become increasingly more popular as part of enhanced surgical recovery programs.9 By providing patients with a higher level of understanding of pain origination and the usual or expected duration and intensity of postsurgical pain, patients can alter their perception of the noxious stimulus and use nonpharmacologic pain management techniques leading to a decreased use of medications. Patients will also be able to identify inappropriate healing, infection, or the development of a chronic pain syndrome by the occurrence of unusual pain intensity or duration.
Acupuncture, as well as acupressure, has been shown to increase the release of inhibitory neurotransmitters such as norepinephrine, serotonin, and enkephalins, thereby enhancing the descending inhibitory pain pathway.10
TENS and stimulation of non-nociceptive receptors at the site of injury with warming and massage rely effectively on “closing the gate” for pain management.
Conclusion
As we strive to utilize a diverse group of pharmacologic and nonpharmacologic pain management modalities with multiple mechanisms of action, a better understanding of the nociceptive process and the actual mechanisms of action of the modalities employed is warranted in order to tailor a regimen for each patient and situation, thereby maximizing the analgesic effect and minimizing complications.
References
- Melzack R, Wall PD. Pain mechanisms: a new theory. Science. 1965;150(3699):971-979.
- Ricciotti E, FitzGerald G. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31(5):986-1000.
- Li H, Xie W, Strong JA, et al. Systemic anti-inflammatory corticosteroid reduces mechanical pain behavior, sympathetic sprouting, and elevation of pro-inflammatory cytokines in a rat model of neuropathic pain. Anesthesiology. 2007;107(3):469-477.
- Chandrasekharan NV, Dai H, Lamar Turepu Roos K, et al. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. PNAS. 2002;99(21):13926-13931.
- Ninkovi J, Roy S. Role of the mu-opioid receptor in opioid modulation of immune function. Amino Acids. 2013;45(1):9-24.
- Schmidt P, Ruchelli G, Mackey SC, et al. Perioperative gabapentinoids: choice of agent, dose, timing, and effects on chronic postsurgical pain. Anesthesiology. 2013;119:1215-1221.
- Premkumar LS, Sikand P. TRPV1: a target for next generation analgesics. Curr Neuropharmacol. 2008;6(2):151-163.
- BJ Anderson, GA Woollard, NH Holford. Acetaminophen analgesia in children: placebo effect and pain resolution after tonsillectomy. Eur J Clin Pharmacol. 2001;57(8):559-569.
- Brown JK, Singh K, Dumitru R, et al. The benefits of enhanced recovery after surgery programs and their application in cardiothoracic surgery. Methodist Debakey Cardiovasc J. 2018;14(2):77-88.
- Kong J, Schnyer R, Johnson K, et al. Complementary and alternative medicine for cancer pain: an overview of systematic reviews. Evid Based Complement Alternat Med. 2013;2013:187-182.
Leave a Reply
You must be logged in to post a comment.