
Associate Professor, Anesthesiology
Wake Forest School of Medicine
Winston-Salem, North Carolina
Wake Forest School of Medicine
Winston-Salem, North Carolina
Dr Bryan and Ms Johnson reported no relevant financial disclosures.
![]() Download the printer-friendly PDF version here.
Download the printer-friendly PDF version here.
Children with rare congenital syndromes tend to undergo multiple diagnostic procedures or surgeries throughout their lifetime that require sedation or general anesthesia.1 Due to the various anatomic and craniofacial features present in children with these syndromes, airway management can be challenging.
Further complicating matters, each syndrome presents its own challenges in terms of airway management: intubation, ventilation, the increased risk for desaturation and/or aspiration, or other non-airway complications.2 Therefore, careful consideration should be given when deciding which airway devices and anesthetic techniques to use during airway management of children with rare diseases.
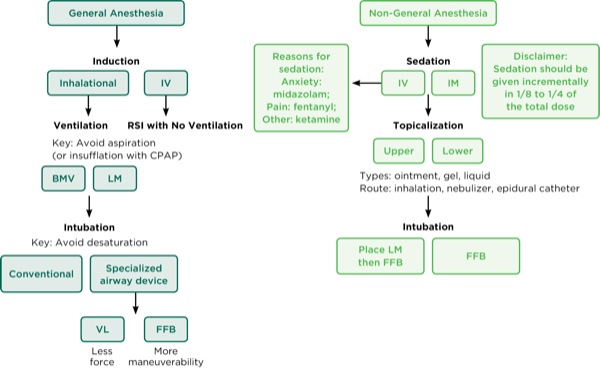
The key to managing these types of cases is identifying where the difficulty in airway management lies and how to address it. There are many alternative means to manage difficult airways in these children. Certain airway and anesthetic techniques may be better alternatives than the initial choice of conventional laryngoscopy and bag-mask ventilation.3
The use of specialized airway devices, such as a video laryngoscope or flexible fiber-optic bronchoscope (FFB), with or without a supralaryngeal device (SLD), may offer a safe alternative as an initial choice.4 The key is to avoid an urgent or emergent intervention, such as performing a surgical technique quickly during a crisis in a patient with limited oxygen reserves.
The options of oral or nasal intubation with an FFB and the use of different SLDs (eg, an LMA ProSeal [Teleflex]; air-Q [Cookgas], etc) for ventilation and/or as a conduit for asleep FFB intubation offer a more controlled setting.
Alternative nonconventional methods adapted from routine clinical care in children are needed.1Depending on the age and cognitive function of the patient, another safe option is the use of “non-asleep” techniques for intubation, for which the patient is either awake or intubated after the administration of minimal to moderate sedation and topical anesthesia of the airway. These techniques allow for spontaneous breathing and preserve muscle tone during airway management.
In order to decide which technique to choose, it is important to consider the many factors involved, such as age; cognitive function; anatomic and physiologic differences, depending on the specific syndrome encountered; and potential reactions or inability to use certain medications and/or anesthetic agents in certain syndromes.2 Regardless of the airway devices used, the goal beyond visualizing the vocal cords or placing the endotracheal tube (ETT) is to minimize complications and safely perform the tasks easily during airway management.
This review discusses 3 separate and rare syndromes in children, Cornelia de Lange syndrome (CdLS), Pitt-Hopkins syndrome (PTHS), and Freeman-Sheldon syndrome (FSS, also called “whistling face” syndrome), as well as their implications for airway and anesthetic management. The clinical features of these syndromes and their implications for airway management as related to intubation, ventilation, and prevention of oxygenation and aspiration are examined.
Case 1: Cornelia de Lange Syndrome
An infant recently diagnosed with CdLS is admitted for further workup at a tertiary care children’s hospital (Figure 1). She underwent a G-tube placement at 6 weeks of age and was reported to be a difficult intubation by her parents, on the basis of experiences during a past operation. She presented for a workup at 3 months of age that would include laryngoscopy, bronchoscopy, and esophagogastroduodenoscopy. The parents ask whether alternative airway techniques may be considered for the patient beyond conventional laryngoscopy and bag-mask ventilation.
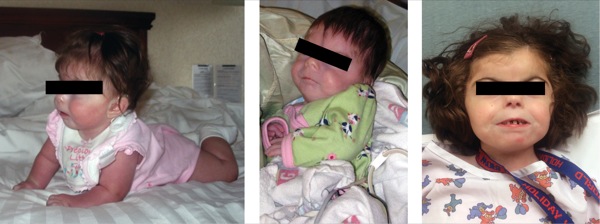
CdLS is a congenital disease affecting approximately 1 in 20,000 births. Clinical symptoms include a depressed nasal bridge, micrognathia, a cleft palate, a small mouth opening, a receding chin, and limited range of motion of the neck. Children affected by this syndrome tend to undergo several procedures in their lifetimes that may require the use of sedation or general anesthesia.
Due to the anatomic abnormalities of the disorder, these children may present with difficult airways. The ability to obtain an adequate seal during bag-mask ventilation may be challenging due to the depressed nasal bridge and receding chin, which may possibly lead to desaturation.
A study of CdLS patients found that approximately 30% of parents and caregivers of pediatric patients reported difficult intubation during past procedures.5 Additionally, 43% reported the need for a smaller ETT.5 Case reports indicate that a variety of airway devices have been used successfully for intubation, and highlight the need for safe techniques (Table 1).
| Table 1. Summary of Literature on Cornelia de Lange Syndrome | ||||
| Authors | Journal; Volume | Year | Title | Main Topics |
|---|---|---|---|---|
| Veall GRQ | Anaesthesia; 49 | 1994 | An unusual complication of Cornelia de Lange Syndrome | A 24-year-old woman with CdLS was undergoing a surgery in which a preoperative anesthesia assessment was impossible due to aggressive behavior and lack of cooperation. Upon extubation, the patient experienced a severe desaturation and required to be intubated and taken to the ICU, where it was discovered that she had congenital heart disease. This would have been valuable information had more thorough preoperative assessments been performed. |
| Munoz Corsini L, De Stefano G, Porras MC, et al | Paediatric Anesthesia; 8 | 1998 | Anaesthetic Implications of Cornelia de Lange Syndrome | Presents a case of a 9-year-old boy who underwent orthopedic surgery. He required a smaller tube than his age usually requires, and after extubation experienced glottal spasm. |
| Fernandez-Garcia R, Perez Mencia T, Gutierrez-Jorda A, et al | Pediatric Anesthesia; 16 | 2006 | Anesthetic management with laryngealmask in childwith Brachmann-de Lange Syndrome | Reports the successful airway management of an 11-year-old child with an LMA after obtaining a grade 4 view with a laryngoscope. |
| August DA, Sorhabi S | Pediatric Anesthesia; 19 | 2009 | Is difficult airway predictable in Cornelia de Lange Syndrome? | Presents a case of difficult intubation in which a 2-year-old girl was able to be oxygenated and ventilated with no issues, but direct laryngoscopy was unsuccessful. Two operators performed direct laryngoscopy using both MAC and Miller devices and were unsuccessful. FFB through an LMA and nasal FFB were also unsuccessful. Finally, a pediatric GlideScope VL was successful and a 4.0-mm ETT was placed. |
| Torres MD, Calvo E, Espla FF, et al | Minerva Anestesiol; 76 | 2010 | Anesthetic management of an adult patient with Cornelia de Lange Syndrome | Short and decreased flexibility of neck, so an awake oral FFB was planned. There were 6 unsuccessful attempts at intubation until a 7.0 ETT was placed. |
| Ingram B, Frost EAM | Middle East Journal of Anesthesiology; 3 | 2011 | Post-operative respiratory complications after palatoplasty in a 19 month oldfemale with Cornelia de Lange Syndrome | Presents a case in which the patient had a previous history of difficult airway. A wis blade was used to intubate and a grade 3 view was obtained, which was improved to a grade 2 view after cricoid pressure was applied. A 4-mm ETT was unsuccessful to place, so a 3.5-mm ETT was used. After extubation, the patient desaturated to the 60s and required an oral airway with CPAP via mask ventilation. |
| Sugiyama T, Okutani R | Anaesthesia; 67 | 2012 | Difficult tracheaintubation in a child with Cornelia de Lange Syndrome using a paediatric Intlock installed in a Pentax Airway Scope | A 7-year-old boy was successfully intubated with a 4-mm ETT using a Pentax Airway Scope with a pediatric intlock placed in it, after unsuccessful direct laryngoscopy due to decreased neck range of motion. |
| Hamilton J, Clement WA, Kubba H | International Journal of Pediatric Otorhinolaryngology; 78 | 2014 | Otolaryngological presentations of Cornelia de Lange Syndrome | Presents 6 cases: 1) 22-year-old man with history of esophageal dismotility and gastroesophageal reflux disease that overspills to larynx with choking attacks. Cases 2-6) laryngomalacia leading to breathing abnormalities and obstructive sleep apnea. |
| Boyle MI, Jespersgaard C, Brondum-Nielsen K, et al | Clinical Genetics; 88 | 2015 | Cornelia de Lange Syndrome | Reviews the clinical presentation of CdLS. Craniofacial considerations: microcephaly, depressed nasal bridge, micrognathia, high and/or cleft palate, thin lips, short neck. Behavioral considerations: attention-deficit issues, hyperactivity, and sleep disturbances. |
| Moretto A, Scaravilli V, Ciceri V, et al | American Journal of Medical Genetics Part C; 172C | 2016 | Sedation and General Anesthesia for Patients with Cornelia de Lange Syndrome: A Case Series | Describes the airway and anesthetic management of 27 patients with CdLS. Six of the 27 patients were intubated, while the others were managed with mask ventilation during the case. Of the 6 that were intubated, 3 were deemed difficult by a grade 3 or 4 view. |
| CdLS, Cornelia de Lange syndrome; CPAP, continuous positive airway pressure; ETT, endotracheal tube; FFB, flexible fiber-optic bronchoscope; LMA, laryngeal mask airway | ||||
CdLS may present prenatally during ultrasound assessment or after genetic testing. Once diagnosed early in infancy, these patients may be especially challenging for airway management due to their anatomic and pathophysiologic differences, leading to severe airway and nonairwayoutcomes, such as cardiac arrest. The goal for airway management in these patients is to safely intubate and ventilate using a specialized airway device, such as an FFB or in combination with an SLD, as the initial technique. Use of this approach may avoid desaturations and other non–airway-related complications.
An elegant technique is to induce the patient, place an SLD to oxygenate and ventilate, and then use that as a conduit to intubate with an FFB, with railroading of an ETT. Additionally, an FFB alone may be used to intubate, or after removal of the SLD.6,7
In this case, after induction, an LMA Classic (Teleflex) was used for oxygenation and ventilation as well as a conduit for intubation using an FFB. This technique was a safe alternative to conventional laryngoscopy, as it was already known that intubation and ventilation were to be challenging from the history provided by the parents (Figure 2).
Case 2: Pitt-Hopkins Syndrome
An 8-year-old boy with PTHS presents for both lower and upper endoscopies (Figure 3). The parent informs you that the last time her child received general anesthesia he became very agitated and aggressive when he awakened. He also was not able to fall asleep for almost 36 hours post-surgery. The parent wants to know whether there is anything that can be done differently to avoid these postoperative complications. What can be done to avoid paradoxical reactions to these medications?

PTHS is a rare genetic syndrome affecting just several hundred patients worldwide. Typical symptoms displayed in children with this syndrome include intellectual disabilities, seizures, hyperventilation, gastrointestinal (GI) issues, and anatomic abnormalities. Intellectual disabilities may make cooperation difficult for the patient and lead to unexpected and severe temperament changes during preoperative assessment.
Additionally, the antiepileptic medications that these children receive may interact with certain medications and anesthetic agents. As for airway management, oxygenation and/or the risk for aspiration are the principal challenges. PTHS children undergo periods of hyperventilation that may lead to low oxygen saturation levels and an increased risk for desaturation during airway management (Table 2). Longer preoxygenation periods before intubation may reduce this risk.
| Table 2. Summary of Literature on Pitt-Hopkins Syndrome | ||||
| Authors | Journal; volume | Year | Title | Main Topics |
|---|---|---|---|---|
| Maini I, Cantalupo G, Turco EC, et al | Journal of Child Neurology; 27(12) | 2012 | Clinical and polygraphic improvement of breathing abnormalities after valproate in a case of Pitt-Hopkins syndrome | A child with PTHS presented with breathing abnormalities consisting of hyperventilation and apnea leading to oxygen desaturation. Diazepam was first used to treat this condition, but did not reduce the episodes and caused excessive sleepiness. Sodium valproate improved saturation levels. |
| Van Balkom IDC, Jelle Vuijk P, FranssensM, et al | Developmental Medicine & Child Neurology; 54 | 2012 | Development, cognition, and behaviour in Pitt-Hopkins syndrome | Ten families were surveyed through a PTHS support group in the Netherlands and Belgium and individuals with PTHS were examined by a psychiatrist. All of the individuals had severe intellectual disabilities with difficulty communicating. |
| Navaratnarajah J | Pediatric Anesthesia; 23 | 2013 | Previously unreported difficult intubation in a child with Pitt-Hopkins syndrome | Case report of a difficult intubation in a PTHS patient. Bag-mask ventilation was easy but required significant insufflation. Direct laryngoscopy only produced a grade III view. The view was improved by adding a roll under the shoulders and applying cricoid pressure. A bougie was then used to successfully intubate the patient. |
| Gaffney C, McNally P | American Journal of Medical Genetics Part A; 167A | 2015 | Successful use of acetazolamide for central apnea in a child with Pitt-Hopkins syndrome | In an awake state, it is common for PTHS children to undergo hyperventilation and breath holding leading to cyanosis. Acetazolamide has been shown to reduce the severity and frequency of these episodes. |
| de Winter CF, Baas M, Bijlsma EK, et al | Orphanet Journal of Rare Diseases; 11 | 2016 | Phenotype and natural history in 101 individuals with PTHS through an internet questionnaire system | Summarizes characteristics of 101 individuals with PTHS. Seizures, hyperventilation, and GI issues seemed to be common problems associated with the syndrome. |
| GI, gastrointestinal; PTHS, Pitt-Hopkins syndrome | ||||
In addition, the high incidence of GI issues may lead to an increased risk for aspiration. However, lower GI problems, such as diarrhea and constipation, tend to outweigh gastroesophageal reflux and upper GI problems.
A recommended technique involving minimal insufflation of the stomach during bag-mask ventilation and the avoidance of positive pressure may be imperative in these patients.8,9 A case report noted a difficult intubation in a child with PTHS, but this is not a general trend in these patients. Their distinctive wide mouth with full lips does not tend to predispose these children to an increased risk for difficult intubation. The main issue tends to be with the anesthetic agents used, as these children often experience paradoxical, adverse effects from different medications.
To further complicate the choice of anesthetic, each child may have differing reactions depending on the specific mutation that led to PTHS.10 Not all patients with this rare syndrome present with difficulties in airway management, and it is important to select the best anesthetic agents to tailor their care. Similarly, in certain syndromes, such as FSS, discussed below, certain medications should not be used, and other more specialized techniques are required.
Case 3: Freeman-Sheldon (“Whistling Face”) Syndrome
A 7-year-old boy was scheduled for orthopedic surgery of the lower extremities (Figure 4). Induction was performed with propofol and fentanyl. For maintenance of anesthesia, remifentanil and propofol were used, and a continuous epidural block was performed for postoperative analgesia.
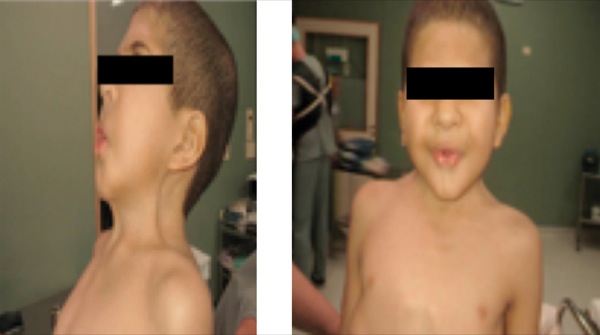
For airway management, a laryngeal mask was used as a guide to introduce the FFB. A metal guide was introduced with a flexible tip, and the FFB and laryngeal mask airway were both withdrawn leaving the guide in place. An ETT was railroaded onto the guide and placed into the trachea. The guide was then removed without difficulty.
FSS is another rare congenital syndrome that presents airway management challenges. It is characterized by several anatomic malformations, especially of the face and extremities. Children with this syndrome tend to have a small mouth with pursed lips, resembling the act of whistling, which gives FSS its informal name. In addition, they often present with a short, webbed neck, joint contracture, and clubbed feet. Due to their musculoskeletal malformations, they often require orthopedic and/or plastic surgery under general anesthesia (Table 3).
| Table 3. Summary of Literature on Freeman-Sheldon Syndrome | ||||
| Authors | Journal; Volume | Year | Title | Main Topics |
|---|---|---|---|---|
| Munro HM, Butler PJ, Washington EJ | Paediatric Anaesthesia; 7 | 1997 | Freeman-Sheldon (whistling face) syndrome. Anaesthetic and airway management | A 2-year-old boy with Freeman-Sheldon syndrome underwent general anesthesia for a surgical procedure. A nasal airway was placed to ventilate. Direct laryngoscopy was the first attempt, but there was inadequate visualization. A size 2 LMA was placed as a conduit for flexible fiber-optic intubation. This technique was successful. |
| Agritmis A, Unlusoy O, Karaca S | Pediatric Anesthesia; 14 | 2004 | Anesthetic management of a patient with Freeman-Sheldon syndrome | Case report of a 19-month-old child with whistling syndrome. She had a flat nose with small nostrils, small mouth opening, micrognathia, and a short neck. After induction with midazolam and sevoflurane, direct laryngoscopy was used to intubate but a grade III view was obtained; the view was improved with cricoid pressure but a 4.0-mm ETT failed to pass. A 3.5-mm ETT was placed. |
| Kim JS, Park SY, Min SK, et al | Pediatric Anesthesia; 15 | 2005 | Awake nasotracheal intubation using fiberoptic bronchoscope in a pediatric patient with Freeman-Sheldon syndrome | Case report of a 6-year-old boy with Freeman-Sheldon syndrome who had limited mouth opening and short, webbed neck. Awake nasotracheal flexible fiber-optic bronchoscopy was performed. The nostrils were topicalized but the larynx was not, which led to difficulty visualizing the vocal cords. A 4.5-mm ETT was successfully inserted. |
| Arora MK, Nagaraj G, Lakhe ST | Anesthesia & Analgesia; 103 | 2006 | Combined spinal-epidural anesthesia for a child with Freeman-Sheldon syndrome with difficult airway | Case report of a 1-year-old child with whistling syndrome who had a small mouth opening, micrognathia and limited neck extension. Spinal-epidural injection of bupivacaine was used to provide surgical anesthesia. |
| Patel K, Gursale A, Chavan D, et al | Indian Journal of Anaesthesia; 57 | 2013 | Anaesthesia challenges in Freeman-Sh?eldonsyndrome | Case report of a 1-year-old child undergoing bilateral herniotomy under anesthesia. An LMA was used to spontaneously ventilate after induction and avoid the use of muscle relaxants. |
| ETT, endotracheal tube; LMA, laryngeal mask airway | ||||
Due to their limited mouth opening and short neck, patients suffering from FSS may be prone to difficult intubation. Several case reports illustrate the need to use an FFB either orally or nasally for intubation.
Of note, these patients are at increased risk for malignant hyperthermia during general anesthesia. For that reason, succinylcholine and halogenated agents should be avoided during airway management.
A “non-asleep” technique, including sedation and topicalization of the airway, may be a good alternative for these patients, as muscle tone is maintained and muscle relaxants may be avoided (Figure 5). First, the patient should be administered an anticholinergic agent, such as glycopyrrolate or atropine, to decrease secretions or maintain heart rate, respectively. Midazolam and ketamine may be used for sedation. Once properly sedated, the oral cavity should be topicalized using either lidocaine gel or ointment, depending on the patient’s age and weight. The gel can be applied to the lips, tongue, and palate using a gloved finger or pacifier. If biting of the scope is an issue, an oral airway placed at a 90-degree angle may prevent further biting.
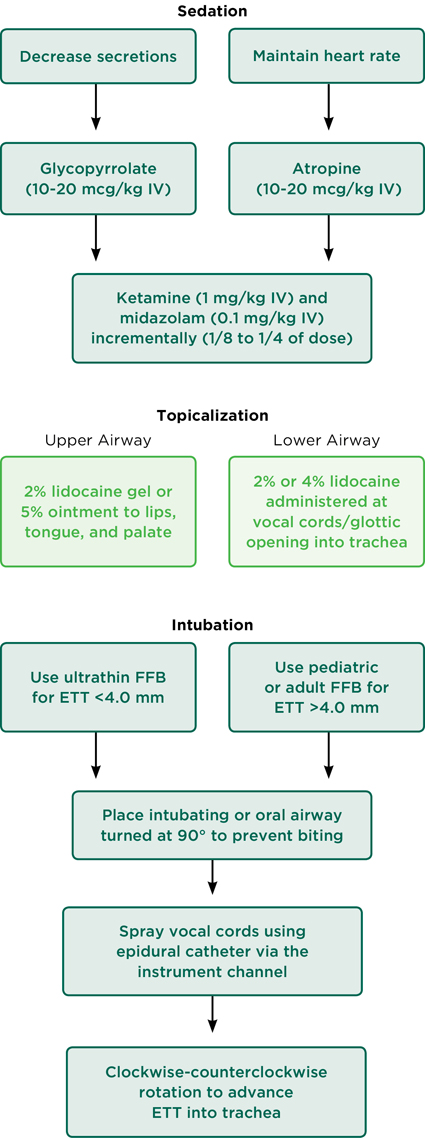
The choice of bronchoscope should be made on the basis of the size of the ETT and the opening of the oral cavity. If the ETT is less than an inner diameter of 4.0 mm, an ultrathin FFB should be used; otherwise a pediatric or adult FFB may be used. An epidural catheter may be placed in the instrument channel of the FFB in order to spray the vocal cords and trachea with lidocaine before passing the ETT.

Once the lower airway has been topicalized, the ETT may be advanced using a clockwise–counterclockwise rotation method. The advantages of this technique are avoidance of inhalational anesthetics, maintenance of muscle control to prevent airway collapse, and enhanced flexibility for placement of the ETT.
Conclusion
Anatomic features in children with rare syndromes may lead to either difficulty visualizing the vocal cords or placing the ETT, or ventilating. The technique used should be tailored to the specific syndrome and the individuals themselves, as there also tends to be significant variation within the same syndrome.
We presented the overall challenges for CdLS, PTHS, and FSS, and the aspects of airway and anesthetic management that are most likely to be difficult (Figure 6). Accordingly, alternative, specialized techniques should be considered in children with these rare syndromes (Figure 7).11,12
Difficult airways in children have been defined using several different criteria. Some syndromes with certain craniofacial features may lead to difficulty visualizing the vocal cords or placing the ETT during direct laryngoscopy. Other syndromes may have features that do not necessarily predispose to difficult intubation but can instead predispose to difficulty with obtaining a seal with a mask, resulting in the need for increasing levels of continuous positive airway pressure (CPAP), leading to insufflation of the stomach and potential aspiration.
Additionally, the prevention of a severe desaturation is an important consideration during airway management, so the relative risk for these events in children with specific rare syndromes should be considered. Finally, many other GI issues, such as delayed gastric emptying or reflux, tend to be more common in certain syndromes, leading to a risk for aspiration beyond the incidence of insufflation during ventilation, as described above.
Potential alternative techniques for CdLS patients include using a laryngeal mask airway for ventilation and placement of a smaller ETT using an FFB for intubation. The focus for PTHS should be minimal use of CPAP during ventilation, or a rapid sequence induction to decrease the risk for aspiration. In patients with FSS, a “non-asleep” method with an FFB and avoidance of succinylcholine and halogenated agents may be appropriate for intubation. Additionally, if there is an adequate mouth opening for the placement of a laryngeal mask airway, then spontaneous ventilation may be preferred for FSS patients, depending on the procedure being performed.
Regardless of the techniques used in children with rare syndromes for intubation and ventilation, the goal is to avoid problems with oxygenation, aspiration, and non-airway complications. More studies will need to focus on which techniques might be adapted from children without syndromes to those with rare ones. Protocols are needed for children with rare syndromes, specifically addressing the problems encountered due to their anatomic and physiopathologic presentations.
The experiences of clinicians, along with the perspectives and perceptions of families of children with these rare syndromes, may assist in providing improved care.
References
- Hosking J, Zoanetti D, Carlyle A, et al. Anesthesia for Treacher Collins syndrome: a review of airway management in 240 pediatric cases. Paediatr Anaesth. 2012;22(8):752-758.
- Heinrich S, Birkholz T, Ihmsen H, et al. Incidence and predictors of difficult laryngoscopy in 11,219 pediatric anesthesia procedures. Paediatr Anaesth. 2012;22(8):729-736.
- Fiadjoe JE, Nishisaki A, Jagannathan N, et al. Airway management complications in children with difficult tracheal intubation from the Pediatric Difficult Intubation (PeDI) registry: a prospective cohort analysis. Lancet Respir Med. 2016;4(1):37-48.
- Fiadjoe JE, Gurnaney H, Dalesio N, et al. A prospective randomized equivalence trial of the GlideScope Cobalt® video laryngoscope to traditional direct laryngoscopy in neonates and infants. Anesthesiology. 2012;116(3):622-628.
- Bryan YF, Kim SM, Johnson KN, et al. Survey of anesthetic and airway management in children and adults with Cornelia de Lange syndrome: parents’ perceptions and perspectives. Anaesth Pain Intensive Care. 2017;21(4):420-426.
- Bryan Y, Chwals W, Ovassapian A. Sedation and fiberoptic intubation of a neonate with cystic hygroma. Acta Anaesthesiol Scand. 2005;49(1):122-123.
- Stricker PA, Budac S, Fiadjoe JE, et al. Awake laryngeal mask insertion followed by induction of anesthesia in infants with Pierre Robin sequence. Acta Anaesthesiol Scand. 2008;52(9):1307-1308.
- Jagannathan N, Sequera-Ramos L, Sohn L, et al. Elective use of supraglottic airway devices for primary airway management in children with difficult airways. Br J Anaesth. 2014;112(4):742-748.
- Templeton TW, Bryan YF. A two-stage approach to induction and intubation of two infants with Pierre Robin sequence using a LMA Classic™ and Air-Q®: two cases report. Korean J Anesthesiol. 2016;69(4):390-394.
- Bryan YF, Johnson KN, McLaughlin DH, et al. Survey of parents’ perception and perspective on airway and anesthetic management in their children with Pitt Hopkins syndrome: Mapping out their clinical care odyssey. Anaesth Pain Intensive Care. 2018. In press.
- Bryan Y, Ovassapian A. Airway management of a 14-year-old obese, mentally retarded male undergoing uvulopalatopharyngoplasty. Acta Anaesthesiol Scand. 2004;48(2):258-259.
- Kirkpatrick K, Ellwood J, Walker RW. Mucopolysaccharidosis type I (Hurler syndrome) and anesthesia: the impact of bone marrow transplantation, enzyme replacement therapy, and fiberoptic intubation on airway management. Paediatr Anaesth.2012;22(8):745-751.
Leave a Reply
You must be logged in to post a comment.